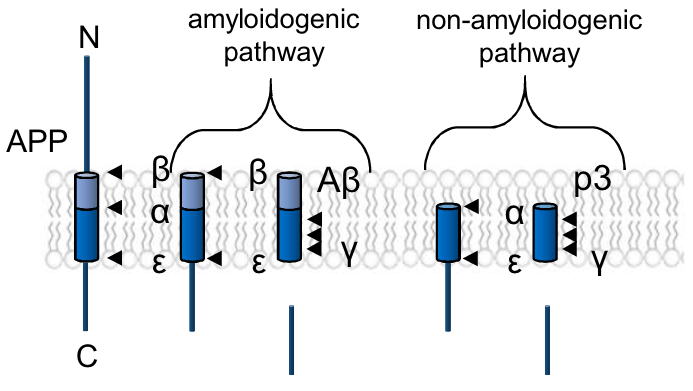
How does the membrane-bound protease presenilin-1 (PSEN1) function in AD? Three recent preprints consider this question.
Harvard researchers found that PSEN1 variants that process the amyloid precursor protein APP into longer amyloid-β (Aβ) proteins in vitro are associated with worse clinical outcomes. Variants with higher Aβ42/Aβ40 ratios in their assay have earlier ages-of-onset, higher levels of amyloid burden and neurodegeneration, and faster rates of cognitive decline. The authors argue that their data support a version of the “amyloid hypothesis,” which places the accumulation of amyloid-β at the heart of AD progression, but acknowledge the need to test whether their PSEN1 variants had issues processing other substrates.
A study from the University of British Columbia supports the need for this qualifying note about other substrates. In a model organism with no equivalent to amyloid-β, the roundworm C. elegans, the authors found that certain PSEN1 mutations tied to familial AD can still cause neurodegeneration. This would suggest that other targets for γ-secretase, the proteolytic complex containing PSEN1, are critical mediators for neurodegeneration in AD.
Previous studies from mice showing neurodegeneration following loss of γ-secretase function have described the resulting pathology as “non-Alzheimer’s” (e.g. Acx et al, 2017) due to a lack of amyloid plaque formation. Our third study, from KU Leuven and UC San Diego, offers insights that could integrate these findings into models of AD. At high concentrations, human Aβ42 inhibits γ-secretase function, impairing processing of both APP and other substrates such as the neurotrophin receptor p75 and Notch. Due to differences in amino acid sequence, this effect is not seen with murine Aβ42.
Taken together, these studies suggest a model in which Aβ42 inhibits γ-secretase, inducing the neurodegeneration seen in numerous studies involving γ-secretase inhibition. In this model, PSEN1 mutations in autosomal-dominant AD contribute disease onset at earlier ages by further biasing the production of Ab in favor of Aβ42, reaching the toxic inhibitory threshold of Aβ42 concentration faster.
The 3 featured studies described here provide exciting new insights into how PSEN1 contributes to AD. In turn, these studies represent a small fraction of the new AD research included in this month’s issue.